
Introduction
Formulation guidance in specialty chemicals refers to the integrated framework of scientific methodology, regulatory compliance, and quality systems that governs how a product—whether a drug, personal care ingredient, polymer, or flavour compound—is designed, tested, and manufactured for consistent performance. Get it right, and brands earn regulatory approval, consistent supply, and customer trust. Get it wrong, and the costs show up as recalls, failed audits, and market access barriers.
Global brands now face audit scrutiny, ingredient transparency demands, and cross-border regulatory complexity at a scale that has no precedent. A 2023 survey by the American Chemistry Council of 58 chemical manufacturers found that 43% reported difficulty obtaining permits, 12% chose not to expand US operations, and 9% moved operations out of the US entirely due to regulatory burden.
The pressure is intensifying at the ingredient level, too. The Modernisation of Cosmetics Regulation Act of 2022 (MoCRA) introduced facility registration, product listing, adverse event reporting, and GMP compliance for cosmetics manufacturers in the US — requirements that push audit scrutiny directly upstream to ingredient suppliers.
Against that backdrop, this blog breaks down the regulatory frameworks — ICH, GMP, 21 CFR — that define acceptable practice, outlines core best practices from preformulation through scale-up, and examines how these standards apply across personal care, flavours and fragrances, and advanced polymers.
Key Takeaways
- Formulation standards are not exclusive to pharma—ICH, GMP, and QbD principles shape quality expectations in personal care, food ingredients, and specialty chemicals.
- Core best practices—preformulation characterisation, excipient compatibility, stability testing, and process control—apply universally across industries.
- Quality by Design (QbD) moves formulation from reactive fixes to structured, risk-based development that cuts late-stage failures.
- Lab-to-commercial scale-up demands a unified quality system to prevent process drift and protect IP.
What Formulation Standards Actually Cover—and Why Industry Context Matters
Formulation is the deliberate combination of active ingredients, functional additives, and excipients into a stable, deliverable product form—whether a tablet, conditioning serum, polymer blend, or flavour emulsion. It's as much a science of materials compatibility as it is of chemistry.
Formulation standards are not a single document but a layered system. Three tiers typically apply:
- International guidelines: ICH series (Q1–Q14 covering stability, analytical validation, and manufacturing)
- National regulations: 21 CFR in the US, BIS in India, EU Cosmetics Regulation
- Internal quality systems: GMP frameworks, SOPs, and batch documentation protocols
Together, these tiers define acceptable development and manufacturing practice.
Pharma vs. Pharma-Adjacent Sectors
The key distinction lies between pharma-regulated formulation—where ICH/GMP compliance is mandatory for regulatory submission—and pharma-adjacent sectors like personal care, flavours and fragrances, and specialty polymers, where the same principles apply as best practice even if not legally mandated in the same way.
Buyers and brand owners in personal care, FMCG packaging, and industrial chemicals now routinely demand GMP-equivalent documentation, batch traceability, and stability data from their formulation partners—regardless of whether their sector mandates it.
Regulatory vs. Best-Practice Standards
A common misconception: "industry standards" in formulation covers both regulatory compliance thresholds (what regulators require) and best-practice benchmarks (what professional formulators do to reduce risk). Distinguishing between the two helps organisations prioritise their quality investments. ISO 22716:2007, for example, provides GMP guidelines for cosmetics and is referenced in FDA's draft cosmetic GMP guidance. It is not legally mandated in every jurisdiction, but brand owners across markets treat it as a baseline expectation.
Key Regulatory Frameworks Every Formulator Should Know: ICH, GMP, and 21 CFR
ICH Q-Series Guidelines
The International Council for Harmonisation (ICH) Q-series provides the global benchmark for pharmaceutical quality:
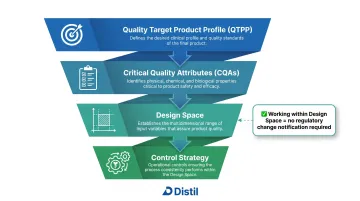
- ICH Q8(R2): Covers pharmaceutical development and design space; defines Quality by Design (QbD), Quality Target Product Profile (QTPP), Critical Quality Attributes (CQAs), and Control Strategy
- ICH Q9: Covers quality risk management principles and tools
- ICH Q10: Covers the pharmaceutical quality system and lifecycle model
- ICH Q7: Covers GMP for Active Pharmaceutical Ingredients (APIs); widely referenced in specialty chemical supply chains beyond pharma

Q7 sets GMP expectations for raw materials and intermediates, making it directly applicable to specialty chemical manufacturers supplying pharma or pharma-adjacent sectors.
GMP vs. cGMP
Good Manufacturing Practice (GMP) is the operational standard governing how formulations are manufactured, controlled, and released—covering equipment qualification, batch records, in-process controls, and deviation management.
cGMP stands for "current" Good Manufacturing Practice, emphasizing that companies must use technologies and systems that are up-to-date, not practices that were standard 10 or 20 years ago. cGMP regulations for drugs are codified at 21 CFR Parts 210 and 211.
21 CFR Relevant Sections
21 CFR (Code of Federal Regulations, Title 21) is the US federal regulatory framework governing food, drugs, and cosmetics. Key parts for specialty chemical formulators include:
- Parts 210/211: Drug manufacturing GMP
- Part 117: Food safety/GMP
- Parts 700–740: Cosmetics labelling, registration, and product warning statements
Formulators targeting the US market need to identify which CFR parts apply to their product category early in development — misclassification is one of the most common causes of regulatory delays at market entry.
Quality by Design (QbD)
ICH Q8(R2) codifies QbD as the principle that quality must be built into a formulation through structured development, not tested into the finished product through end-point inspection. Structured development means defining a Quality Target Product Profile (QTPP), identifying Critical Quality Attributes (CQAs), and establishing a Design Space before scale-up begins.
The operational payoff is twofold:
- Lower non-conformance costs: defects are caught at design stage, not during batch release
- Regulatory flexibility: working within an approved design space is "not considered a change," eliminating post-approval submission requirements
Regional Equivalents
- EU Cosmetics Regulation No. 1223/2009: Governs finished cosmetic products, with recent amendments including Regulation (EU) 2024/996 (Vitamin A, Alpha-Arbutin, endocrine-disrupting substances) and Regulation (EU) 2023/1545 (fragrance allergen labelling)
- FSSAI regulations: Govern food ingredients and flavours in India; the Food Safety and Standards (Food Products Standards and Food Additives) Regulations, 2011 were updated in January 2024
- REACH: EU Regulation on Registration, Evaluation, Authorisation and Restriction of Chemicals requires registration of substances manufactured or imported in quantities of 1 tonne or more per year per company
Core Formulation Best Practices Across the Development Cycle
Preformulation Characterisation
Preformulation characterisation maps the physicochemical properties of the active ingredient before any excipient selection or dosage form decision is made. The properties that matter most:
- Solubility profile across pH ranges
- Particle size and polymorphism
- Hygroscopicity
- Thermal stability
ICH Q8(R2) states that pharmaceutical development should include studies on "physicochemical and biological properties of the drug substance" and that "critical formulation attributes and process parameters are generally identified through an assessment of the extent to which their variation can have impact on the quality of the drug product."
What happens when this step is skipped:
- Incompatibility failures during stability testing
- Reformulation costs and project delays
- Scale-up surprises due to unforeseen material behaviour
Excipient Selection and Compatibility
Every inactive ingredient — binder, emulsifier, stabiliser, solubiliser, preservative — must be selected for its specific technical role and tested for compatibility with the active and all other excipients in the system. ICH Q8(R2) requires justification of excipient choice and concentration ranges.
Compatibility testing protocols: These stress conditions — temperature, humidity, light, pH extremes — accelerate degradation and expose interaction risks before you commit to a formulation design. Results feed directly into shelf-life claims and packaging specifications. Yu et al. (2014) note that "systematic drug-excipient compatibility studies minimise unexpected stability failures which usually lead to increased development time and cost."
Stability Testing and Shelf-Life Determination
ICH Q1A(R2) is the industry benchmark for establishing shelf-life claims — and while originally designed for pharmaceutical drug substances, its conditions are now standard practice in personal care and specialty chemical formulations.
Standard ICH accelerated stability conditions:
- Accelerated: 40°C ± 2°C / 75% RH ± 5% RH for 6 months
- Long-term: 25°C ± 2°C / 60% RH ± 5% RH for 12 months
Testing frequency: every 3 months in year 1, every 6 months in year 2, annually thereafter. A "significant change" at accelerated conditions is defined as a 5% change in assay from initial value.

Process Controls and In-Process Testing
A formulation isn't complete without defined Critical Process Parameters (CPPs) and in-process controls that ensure every batch meets specification. Per ICH Q8(R2), a CPP is "a process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality."
In-process checkpoints that prevent batch-to-batch variability during scale-up:
- Viscosity — detects mixing or temperature deviations early
- pH — confirms ionic environment and preservative efficacy
- Particle size — critical for emulsions, dispersions, and UV filter suspensions
- Assay — verifies active concentration against specification
QbD, Critical Quality Attributes, and Control Strategy
Critical Quality Attributes (CQAs)
CQAs are the measurable physical, chemical, biological, or microbiological properties that must fall within a defined range to ensure the product performs as intended. Defining them early shapes every downstream formulation and testing decision — from excipient selection to release specifications. Common CQA categories include:
- Physical: particle size, viscosity, dissolution rate
- Chemical: assay, degradation products, pH
- Biological: potency, sterility, endotoxin levels
- Microbiological: bioburden, preservative efficacy
Design Space
ICH Q8(R2) defines Design Space as "the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality. Working within the design space is not considered as a change."
This gives manufacturers operational flexibility: variations within the design space avoid regulatory change notifications, while movement outside triggers post-approval change processes.
Control Strategy
A Control Strategy ties CQAs, process parameters, and input material controls into a documented quality system. ICH Q8(R2) defines it as "a planned set of controls, derived from current product and process understanding that ensures process performance and product quality."
In practice, this means quality is built into the process rather than tested in at the end — reducing batch failures and post-approval surprises. Yu et al. (2014) state that "the goals of implementing pharmaceutical QbD are to reduce product variability and defects, thereby enhancing product development and manufacturing efficiencies."
Applying Formulation Standards in Personal Care, F&F, and Advanced Polymers
Each of Distil's three core verticals applies formulation standards differently. Here is how those requirements take shape in practice.
Personal Care
Formulation standards translate into personal care through:
- Stability testing for emulsions and actives
- Preservative efficacy testing aligned with ISO 11930:2019 ("Cosmetics—Microbiology—Evaluation of the antimicrobial protection of a cosmetic product")
- Skin compatibility and compliance with ingredient restrictions under the EU Cosmetics Regulation and India's Drugs and Cosmetics Act
Brand owners now expect GMP-aligned manufacturing and full batch traceability from their ingredient suppliers and contract formulators. Distil's R&D team — with direct experience from L'Oréal, BASF, and Dow — applies this compliance knowledge to application-specific formulation development and ingredient trials.
Flavours and Fragrances
Formulation considerations specific to F&F include:
- IFRA (International Fragrance Association) standards for safety and allergen limits; the IFRA Standards Library represents the global benchmark for fragrance ingredient safety
- FEMA GRAS status for flavour ingredients; the FEMA Expert Panel has evaluated approximately 2,200 flavouring substances since 1959
- Encapsulation and delivery system stability: solubility and volatility characteristics govern formulation choices in ways distinct from solid dosage forms
Advanced Polymers
Polymer formulation demands tight control at both the material and process levels:
- Rheology characterisation: Melt flow index (MFI) consistency per ASTM D1238-23a and ISO 1133-1:2022
- Additive compatibility: Stabilisers, plasticisers, flame retardants — each with defined loading ranges per end-use
- Process parameter control: Temperature and shear profiles directly determine final material properties
Automotive, wire and cable, and packaging sectors all require documented formulation specifications and batch-level traceability as a supplier qualification condition.

IATF 16949:2016 extends to bulk chemical suppliers, with AIAG's CQI-37 manual addressing safety-critical bulk materials and D-22 governing formulation ingredient changes.
From Lab to Commercial Scale: Turning Formulation Rigour into Practice
The Tech Transfer Challenge
The key challenge of tech transfer is translating a formulation that performs at lab scale (grams to kilograms) into a commercially reproducible process at industrial scale without drift in quality attributes. This covers equipment differences, batch size effects on mixing and heat transfer, and the importance of scale-independent process parameters.
The ISPE Good Practice Guide: Technology Transfer (3rd Edition, 2018) aligns with ICH Q8, Q9, Q10, Q11 and FDA's lifecycle approach to process validation, providing the authoritative template for specialty chemical scale-up documentation.
Unified Quality System
A unified quality system prevents process drift across a manufacturing partner network through:
- Standardised SOPs
- Batch record templates
- Deviation management
- Shared analytical methods
This ensures the approved formulation design is faithfully reproduced regardless of which facility executes the batch. Distil operates this model across its 20+ manufacturing partner network: real-time production visibility, a centralised quality layer, and consistent standards at every site. The result is commercial-scale production without capital expenditure, with formulation IP remaining under the brand's control.
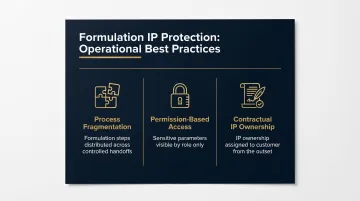
IP Protection as Formulation Governance
IP protection is a formulation governance issue, not just a legal one. Operational best practices include:
- Process fragmentation: Distributing formulation steps across controlled handoffs so no single partner holds the complete picture
- Permission-based access: Limiting formulation data visibility by role, so sensitive parameters stay protected even within production environments
- Contractual IP ownership frameworks: Establishing from the outset that formulation IP belongs to the customer, not the manufacturer
These protections should be embedded in any external manufacturing arrangement before production begins. Trade secret law — one of the oldest IP mechanisms in chemical research — covers formulas, processes, and proprietary methods, but only when backed by operational controls that enforce confidentiality in practice.
Frequently Asked Questions
What is the definition of a formulation?
A formulation is the deliberate combination of an active ingredient with functional additives and excipients into a stable, deliverable product form. The specific components and their ratios are engineered to achieve a defined performance outcome, whether therapeutic, cosmetic, or functional.
What is the ICH Q7 guideline?
ICH Q7 is the international GMP guideline specifically for Active Pharmaceutical Ingredients (APIs), covering manufacturing controls, quality systems, and documentation requirements. It is widely referenced in specialty chemical supply chains as a benchmark for API-adjacent ingredient manufacturing.
What is 21 CFR and GMP?
21 CFR (Title 21 of the US Code of Federal Regulations) is the federal legal framework governing food, drugs, and cosmetics in the US. GMP (Good Manufacturing Practice) is the operational quality standard within 21 CFR, defining how products must be manufactured, tested, and documented to ensure safety and consistency.
What is PDR in pharma?
PDR stands for Physicians' Desk Reference, a compendium of prescribing information for approved drug products. In a formulation context, it is referenced to verify approved dosage forms, ingredient listings, and labelling standards for commercially marketed products.
What is Quality by Design (QbD) in formulation?
QbD is a systematic development approach, codified in ICH Q8, where quality is built into a formulation from the start by defining target performance outcomes, identifying critical quality attributes, and establishing a scientifically justified design space—rather than relying on end-product testing alone.
What regulatory standards govern personal care product formulation?
Personal care formulation is governed by regional regulations including EU Cosmetics Regulation No. 1223/2009, India's Drugs and Cosmetics Act, and FDA 21 CFR Part 700–740, alongside industry standards such as ISO 11930 (preservative efficacy) and IFRA fragrance safety guidelines. GMP-aligned manufacturing is increasingly expected by brand owners even where not legally required.


